國際檢查動態之FDA對生物等效性研究實施有因檢查的原因分析
為了探討FDA對仿制藥生物等效性研究工作涉及的臨床、分析及溶出研究單位實施“有因”檢查的最常見原因,并且對此類檢查的結果進行匯總,申辦者向美國食品藥品監督管理局(FDA)藥品審評和研究中心(CDER)遞交藥物生物等效性(BE)研究,以支持簡化新藥申請(ANDAs)和批準后補充申請。據此,環球網校分享國際檢查動態之FDA對生物等效性研究實施有因檢查的原因分析供大家了解。
1.前言
申辦者向美國食品藥品監督管理局(FDA)藥品審評和研究中心(CDER)遞交藥物生物等效性(BE)研究,以支持簡化新藥申請(ANDAs)和批準后補充申請。為了保證仿制藥與相應的參比制劑(RLD)在治療方面等效,仿制藥必須與參比制劑藥學等效和生物等效。
生物等效性定義為:在合理設計的研究中,藥學等效制劑或可替換藥物在相似條件下以相同摩爾劑量給藥后,其活性成分或活性分子到達藥物作用部位時的速度和程度沒有顯著性差異。有多種體內和體外的方法驗證生物等效性。其中,體內藥代動力學生物等效性研究是最常用的比較受試制劑與參比制劑的系統暴露特征的方法。ANDA生物等效性申報資料中,包含了四個主要的研究報告:生物分析方法、臨床研究報告、統計分析和體外溶出試驗。在生物等效性研究結果的審評過程中,相關的缺陷會被指出并建議申辦者更正。如果在生物等效性研究審評過程中發現有任何真實信息顯示存在學術行為不端、嚴重違反受試者保護或者影響生物等效性數據的,FDA審評員將會要求FDA科學調查辦公室(OSI)對生物等效性研究的溶出、分析和/或臨床機構實施“有因”檢查。應此要求,OSI將會安排日程對有違規嫌疑的機構進行檢查。OSI實施檢查是為了保證人類受試者的權利、安全及權益能夠得到保護,并核查生物等效性研究是否符合美國聯邦法規(CFR)第21章第320節生物利用度與生物等效性的要求。此程序有助于保證遞交給FDA的生物等效性研究資料的完整性和可靠性。
本研究的目的是為了探討FDA對仿制藥生物等效性研究工作涉及的臨床、分析及溶出研究單位實施“有因”檢查的最常見原因,并且對此類檢查的結果進行匯總。我們期望通過發表本研究中列出的問題,會有助于制藥企業發現仿制藥生物等效性研究不合規的根本原因,以避免此類問題再次發生,從而促進僅僅高質量的、安全的、有效的仿制藥獲得批準。
環球網校友情提供:執業藥師考試論壇,隨時與廣大考生朋友們一起交流!
編輯推薦:
國家食藥監管理局發布化學藥生物等效性(BE)試驗備案范圍和程序
12月1日起化學藥生物等效性(BE)試驗由審批制改為備案管理
為了探討FDA對仿制藥生物等效性研究工作涉及的臨床、分析及溶出研究單位實施“有因”檢查的最常見原因,并且對此類檢查的結果進行匯總,申辦者向美國食品藥品監督管理局(FDA)藥品審評和研究中心(CDER)遞交藥物生物等效性(BE)研究,以支持簡化新藥申請(ANDAs)和批準后補充申請。據此,環球網校分享國際檢查動態之FDA對生物等效性研究實施有因檢查的原因分析供大家了解。
2.方法
通過對FDA內部數據庫的檢索,確定了從2003年1月至2011年12月(共9年)進行的與ANDA生物等效性研究相關的“有因”檢查的原因。這些申請包含了包括固體口服制劑、藥物透皮遞釋系統、咀嚼藥物及口服混懸劑或腸外給藥制劑等多種劑型的生物等效性試驗。并按照以下方面對“有因”檢查進行分析:(1)檢查的原因分類;(2)被檢查單位的類型,如臨床、分析及溶出研究單位;(3)每年要求的檢查次數,以及(4)檢查結果。
要求實施“有因”檢查的常見原因按下列標準進行分類(表1):
2.1數據可靠性和有效性問題
此類型問題可進一步細分為以下具體原因(表2):
a.要求實施檢查以確保數據的準確性:懷疑研究結果存在偏倚,可能會導致生物等效性結論無效。
b.申辦者遞交的信息與FDA獲得的其他信息存在分歧:FDA審評員發現公司遞交的生物等效性研究結果與FDA獲得的其他數據(如藥代動力學參數或溶出結果)相比較時,存在顯著性差異,并且此類差異并不合理。
c.研究方案偏離:公司沒有保證研究按照研究計劃執行。
d.方法驗證不充分:研究缺乏足夠的驗證研究數據,例如沒有進行交叉驗證研究。
e.遞交的信息不一致或相互矛盾:審評員發現公司報告中存在無法解釋的不一致之處。
2.2再分析的樣本量過多
在生物等效性研究中,樣本進行再分析的比例很高,并且申請人無法提供令人信服的依據支持再分析的合理性。
2.3被檢查單位此前有不良檢查記錄
此前對該單位檢查時發現在該單位進行的研究存在數據可靠性方面的問題。要求此次檢查的目的是確定正在審評的在該單位進行的研究是否也采用了類似的不規范研究操作。
2.4文件不完整
公司并沒有保留足夠的、準確的對確定生物等效性至關重要的文件。
2.5研究設計與實施不當
此類型的實例包括:再次給藥研究中對照受試者樣本量不足或者分析研究中質控濃度的選擇不當。
2.6“其他”
不屬于上述列出類型的其他缺陷。例如:與出現嚴重不良事件和/或研究驗證方面的問題。
表1. FDA從2003到2011年要求實施“有因”檢查的原因分類
|
原因分類 |
要求檢查數 |
百分比, % |
|
|
1 |
數據可靠性與有效性問題 |
55 |
60.4 |
|
2 |
再分析的樣本量過多 |
7 |
7.7 |
|
3 |
被檢查單位此前有不良檢查記錄 |
11 |
12.1 |
|
4 |
文件不完整 |
9 |
9.9 |
|
5 |
研究設計與實施不當 |
4 |
4.4 |
|
6 |
“其他” |
5 |
5.5 |
|
合計 |
91 |
100 |
|
表2. 數據可靠性與有效性問題方面的具體原因
|
具體原因 |
要求檢查數 |
百分比,% |
|
|
1.1 |
要求實施檢查以確保數據的準確性 |
34 |
61.8 |
|
1.2 |
申辦者遞交的信息與FDA獲得的其他信息存在分歧 |
2 |
3.6 |
|
1.3 |
研究方案偏離 |
5 |
9.1 |
|
1.4 |
方法驗證不充分 |
8 |
14.5 |
|
1.5 |
遞交的信息不一致/相互矛盾 |
6 |
10.9 |
|
合計 |
55 |
100 |
|
環球網校友情提供:執業藥師考試論壇,隨時與廣大考生朋友們一起交流!
編輯推薦:
國家食藥監管理局發布化學藥生物等效性(BE)試驗備案范圍和程序
12月1日起化學藥生物等效性(BE)試驗由審批制改為備案管理
為了探討FDA對仿制藥生物等效性研究工作涉及的臨床、分析及溶出研究單位實施“有因”檢查的最常見原因,并且對此類檢查的結果進行匯總,申辦者向美國食品藥品監督管理局(FDA)藥品審評和研究中心(CDER)遞交藥物生物等效性(BE)研究,以支持簡化新藥申請(ANDAs)和批準后補充申請。據此,環球網校分享國際檢查動態之FDA對生物等效性研究實施有因檢查的原因分析供大家了解。
3.結果與討論
檢索到2003年1月到2011年12月 FDA 對ANDA實施了累計90項“有因”檢查。這些檢查涉及66個ANDA申請。在此期間,FDA共受理了7,207個ANDA申請。因此,2003年到2011年期間,被要求實施“有因”檢查的比例占所有ANDA申請的0.9%。需要說明的是,除“有因”檢查外,FDA還會實施“常規”檢查,該檢查可在沒有發現行為不端的情況下啟動或者作為投訴的跟蹤。通常,“常規”檢查用于對支持注冊申請,如新藥上市申請、ANDA及新藥臨床申請等開展研究的評估。對“常規”檢查的評估不屬于本文所探討的范疇。
在通常情況下,一項典型的藥代動力學生物等效性研究的參與者包含受試者服藥和血樣采集的臨床研究單位和對血樣進行分析以測定藥物濃度的分析單位。
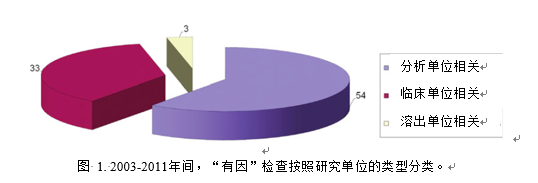
同時臨床終點生物等效性研究可能會涉及多家臨床研究單位。對于某些劑型,如片劑或膠囊,ANDA申辦者也會進行比較溶出試驗為生物等效性提供支持。在90項“有因”檢查中,有54項涉及分析單位的檢查(圖1)。此類檢查主要是針對所涉及的生物分析方法的準確性與可靠性。另有33項涉及臨床研究單位的檢查。此類檢查旨在確定臨床研究的開展是否合規,受試者的權利、安全及權益是否得到了保護。有3項檢查涉及溶出研究單位。要求對溶出單位實施檢查的原因是FDA審評員發現ANDA申辦者遞交的溶出結果與監管部門從其他申請資料掌握的參比制劑溶出的信息存在差異,甚至ANDA申辦者的溶出試驗報告本身就存在不一致。
我們隨后對啟動“有因”檢查的常見原因進行了分析。通過研究發現,FDA要求實施“有因”檢查最常見的原因與“數據可靠性和有效性問題”相關,占要求實施檢查總數的60.4%(表1)。位列第二的要求實施“有因”檢查的最常見原因是被檢查單位此前有不良檢查記錄。這種情況下,FDA審評員要求檢查的目的是為了核實以往對該單位檢查時發現的數據可靠性類似不良問題在正在審評的研究中仍然存在。該原因占要求實施檢查總數的12.1%。“文件不完整”也屬于嚴重的問題,占檢查總數的9.9%。例如,如果申辦者沒有保留研究受試者充分且準確的病歷,并且FDA審評員確認此類數據不完整可能影響研究的結果,審評員可能要求實施檢查以便澄清及確認。除上述最常見的三種原因外,我們對其他導致“有因”檢查的原因也進行了分析。此類原因與申請材料中發現的研究設計、研究實施、標準規程、數據報告及樣本再分析缺陷有關(見表1)。對于無法明確歸類的原因,我們將其列為“其他”類。屬于此類型的“有因”檢查包括一例受試者在空腹生物等效性研究中死亡的病例。FDA審評員對申辦者的研究方案及醫學記錄進行檢查,并未發現有嚴重疏忽或醫療管理不善的證據。但是審評員對于死亡事件及嚴重不良事件仍表示擔憂。因此,為了進一步核實臨床試驗和醫學記錄、研究者的資質以及所用標準操作規程(SOPs)對評價和管理受試者安全問題和不良事件的適用性,審評員要求對其實施“有因”檢查。
表3. FDA要求實施“有因”檢查的原因舉例
|
“有因”檢查的原因 |
實例 |
|
|
1 |
數據可靠性和有效性問題 |
見表4 |
|
2 |
再分析的樣本量過多 |
在空腹及進食研究中,大量樣本分析批被中斷和/或分析物被重分析;申辦者對中斷及重分析沒有提供充分的依據。此外,還有大量的樣本中分析物重新積分,但是申辦者沒有對重新積分提供充分的依據。 |
|
3 |
被檢查單位此前有不良檢查記錄 |
此前在對該研究單位進行檢查時發現許多受試者研究樣本都存在完整性的問題。 |
|
4 |
文件不完整 |
申辦者在研究受試者的進度記錄本上沒有保留充分且準確的病案歷史。 |
|
5 |
研究設計與實施不當 |
再次給藥研究中對照受試者樣本量不足,在實施異常值檢測時缺乏可用的標準操作規程。此外,分析方面的缺陷還包括了質控濃度選擇不當。 |
|
6 |
“其他” |
要求對臨床機構進行檢查是基于已知特定藥物引起嚴重不良事件以及研究受試者住院。 |
表4. 因“數據可靠性和有效性問題”而要求實施“有因”檢查舉例
|
“有因”檢查的原因 |
實例 |
|
|
1.1 |
要求實施檢查以確保數據準確性 |
要求實施檢查以核實受試者X在生物等效性研究中所有采樣時間點所測得的藥物濃度都低于定量下限;核實公司規程在研究單位是否得到嚴格執行以確保受試者用藥劑量,并且確認不存在其他可導致生物等效性研究結果無效的分析缺陷。 |
|
1.2 |
申辦者與FDA內部數據存在分歧 |
分析物的AUC0-t、AUC∞和Cmax等參數值與FDA從其他內部獲得的數據存在較大的偏離。 |
|
1.3 |
研究方案偏離 |
申辦者沒有確保研究按照研究計劃執行。 |
|
1.4 |
方法驗證不充分 |
對于所關注的分析物缺少交叉驗證的研究數據 |
|
1.5 |
提交資料中信息不一致/相互矛盾 |
申辦者提供的研究日期。自相矛盾,并對受試者樣本的儲存時間和穩定性產生影響。 |
我們進一步對“數據可靠性和有效性”方面的原因進行了具體的分析。在此類別中,最常見的原因是為了核實申辦者遞交的生物等效性研究的數據是否準確。例如,生物等效性審評員注意到某例受試者在生物等效性研究中所有采樣時間點所測得的藥物濃度都低于定量下限。隨即要求對其實施檢查,以核實臨床單位是否嚴格執行了公司的規程以確保受試者的用藥劑量,并且核實不存在其他可導致生物等效性研究結果無效的分析缺陷。“方法驗證不充分”是此類型中第二大常見問題。此外,如果FDA審評員發現申辦者遞交的材料中有信息不一致或相互矛盾且無合理解釋的,則會要求對其實施檢查。除上述提到的原因外,還有小部分檢查則是基于潛在的不合規問題和不足,例如:研究方案偏離,研究者提供的信息與FDA獲得的其他信息存在無法解釋的不一致(見表2)。
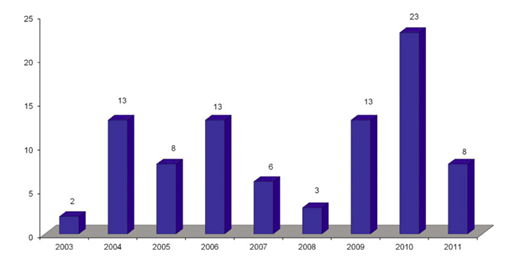
圖2. 2003年到2011年每年要求“有因”檢查的數量
需要說明的是,本次研究中提及的多項檢查并非是因為單一的問題而要求實施檢查的。在通常情況下,由于研究報告中出現的多種問題而暴露的系統性問題,很可能會引起FDA審評員對研究是否有效的擔憂,從而啟動“有因”檢查。例如,FDA審評員發現相同樣本因為不同的原因多次重復檢測,并且申辦者對此類重測未提供充分的依據。此外,FDA審評員在審閱分析報告細節時,發現申辦者提供的分析研究存在明顯的不一致、不相關以及無效的問題。綜合上述所有原因,審評員做出了要求“有因”檢查的決定。表III和表IV呈列了FDA審評員要求實施“有因”檢查的原因的某些典型案例。
對于每年ANDA申請中要求實施“有因”檢查的數量,并沒有明顯的趨勢。2003年,僅要求2項“有因”檢查,但在2010年,則實施了23項“有因”檢查(圖2)。在9年研究期間,平均每年要求10項“有因”檢查。
檢查結果分為以下三種:無需采取措施(NAI),自愿采取措施(VAI)以及采取官方措施(OAI)。NAI是指在檢查時沒有發現違規情況或操作,或者違規情況并不需要采取進一步的監管措施。VAI是指在檢查中發現有偏離法規的情況或操作,但是申請人隨即采取自愿措施對此類情況或操作進行了整改。OAI則是指檢查時發現違規情況/操作,建議采取監管和/或管理方面的措施。如圖3所示,在本研究所考察的“有因”檢查中,有15項屬于NAI,有53項屬于VAI,而15項則屬于OAI。圖3還顯示,有7項尚未得出結論的檢查。從這些數據可以看出,FDA審評員要求實施的“有因”檢查中,有75%的檢查會被合規辦公室定性為存在潛在問題(VAI)或嚴重問題(OAI)。此類生物等效性研究將要求申辦者采取相應的官方或自愿的糾正措施。

圖 3. 從2003年到2011年“有因”檢查結果的類型
環球網校友情提供:執業藥師考試論壇,隨時與廣大考生朋友們一起交流!
編輯推薦:
國家食藥監管理局發布化學藥生物等效性(BE)試驗備案范圍和程序
12月1日起化學藥生物等效性(BE)試驗由審批制改為備案管理
為了探討FDA對仿制藥生物等效性研究工作涉及的臨床、分析及溶出研究單位實施“有因”檢查的最常見原因,并且對此類檢查的結果進行匯總,申辦者向美國食品藥品監督管理局(FDA)藥品審評和研究中心(CDER)遞交藥物生物等效性(BE)研究,以支持簡化新藥申請(ANDAs)和批準后補充申請。據此,環球網校分享國際檢查動態之FDA對生物等效性研究實施有因檢查的原因分析供大家了解。
4.結論
本項研究探討了FDA 對參與ANDA生物等效性研究的臨床研究單位、分析研究單位及溶出研究單位實施“有因”檢查的常見原因。導致采取官方及自愿糾正措施結果的檢查可能會延長申辦者的申請獲得批準的時間,因此應當予以避免。希望通過此文章的發表可以為申請人在力爭符合FDA法規方面提供方法,從而能夠促進安全有效的藥物更快地獲得批準。
(來源:Li, B.V., Davit, B.M., Lee, C.H. et al. AAPS J , 2013 Jan, 15(1): 10-4,作者:美國CDER仿制藥辦公室李冰等,翻譯:王安娜,審校:董江萍)
原文刊登于《國際藥品檢查動態研究》第2卷第1期(總第4期),2017,P20
環球網校友情提供:執業藥師考試論壇,隨時與廣大考生朋友們一起交流!
編輯推薦:
國家食藥監管理局發布化學藥生物等效性(BE)試驗備案范圍和程序
12月1日起化學藥生物等效性(BE)試驗由審批制改為備案管理
最新資訊
- 天津市藥品監督管理局關于啟用審批專用章等新印章的公告2025-11-03
- 上海藥監局:《上海市第二類創新醫療器械特別審查程序》發布2025-09-11
- 國家藥監局公布:2025年8月全國執業藥師注冊情況2025-09-10
- 江蘇藥監局10月1日前必須完成新版許可證申領政策解析2025-08-29
- 國家藥監局公布:2025年7月全國執業藥師注冊情況2025-08-28
- 錯過執業藥師繼續教育怎么辦?別慌,這些省份可補學!2025-08-06
- 人社局明確:執業藥師對應中級職稱,執業藥師含金量升級!2025-07-29
- 藥監局公布!執業藥師注冊人數驟降超1萬2025-07-24
- 遼寧省發布2025年執業藥師繼續教育學習安排2025-07-16
- 2025年湖北執業藥師繼續教育報名中,11月30日截止2025-07-16
 打卡人數
打卡人數


